一、背景
本人邹俊岗,于 2018 年 4月向哈佛大学医学院 Gil Alterovitz 教授(http://www.mit.edu/~gil/)申请,后成为其领导的生物医学控制实验室(https://projects.iq.harvard.edu/bcl)的本科科研实习生。经过与教授商定,我的实习时间为2018年7月1日至2018年12月31日,主要参与教授的FHIR Genomics项目,在其中使用软件工程的方法完成FHIR Genomics Convertor项目。
根据同Gil教授的商议,本人参与其每周两次的国际会议,并接受教授的指导和监督,整个科研实习的进度和流程会被严格监控。就整个科研流程而言,前期会进行生物信息理论的相关学习,之后会使用软件工程的方法搭建FHIR Genomics Convertor软件,最后在进过设计、编码、测试、发布的阶段后完成FHIR Genomics Convertor项目。
二、项目内容
(一)项目背景介绍
- FHIR Genomics
(1)FHIR Genomics介绍
FHIR Genomics标准是访问和交换复杂基因组数据的可行且有效的标准。 FHIR Genomics的初步试验于2016年1月的FHIR Connectathon进行。 通过努力实现Connectathon提出的用例,测试FHIR Genomics标准带来的优势。 FHIR Genomics标准仍处于开发阶段,可在https://projects.iq.harvard.edu/fhirgenomics上查看项目的进度变化。
FHIR Genomics是HL7 FHIR临床数据标准的扩展。它解决了下一代基因组测序(NGS)的标准化问题,这对于医生,遗传咨询师和其他医疗服务提供者来说是非常非常重要的,可以通过这个标准为病人提供知识密集型的个性化护理。 同时,FHIR Genomics小组还建立了FHIR Genomics Reference服务器,该服务器配备了SMART技术(用于支持可替代医疗应用的医疗保健标准),用于临床基因组学应用的原型设计。这样的程序必须能够按需获取患者的基因组数据测试结果(“test once”),以交叉引用一个或多个临床基因组学知识库(“query often”)。
(2) FHIR Genomics Convertor
在各个行业都是有行业标准的,这样才能统一规范而方便后面的分析,在生物信息学领域中主要是各种大量序列数据、注释数据等,这些都是有特定的格式去表示,很多格式有广泛的使用,如fastq(https://en.wikipedia.org/wiki/FASTQ_format)、VCF(http://samtools.github.io/hts-specs/VCFv4.2.pdf)、SAM(http://bio-spring.info/sam-format/)等等。
FHIR Genomics标准设计的初衷是为访问和交换基因组数据提供可行且有效的标准。而在现有的医疗资源的交换过程中存在如疾病名称不统一等情况,而多样的格式也阻碍了医疗资源的交换。因此,我们为FHIR Genomics标准实现一个软件FHIR Genomics Convertor,用于解析fastq文件、VCF文件、SAM文件等等,并将其存储在服务器的数据库中。之后按照用户的需求,对基因数据进行寻找,基于FHIR的查询,将指定的数据转换成FHIR Genomics标准,并上传至用户的HAPI Server中。
2. 项目环境
(1)前端
HTML+CSS+JS(angularjs框架)
(2)Web服务器
Nginx
(3)后端
Node.js(Express框架)实现服务的响应
Python脚本实现对特定文件的解析
(4)数据库
前期使用非关系型数据库MongoDB
后期会增加对HDFS、MySQL等数据库的支持。
(二) 项目实现
在明确了需求之后,我在第一周对系统进行了概要设计,系统架构如图所示:
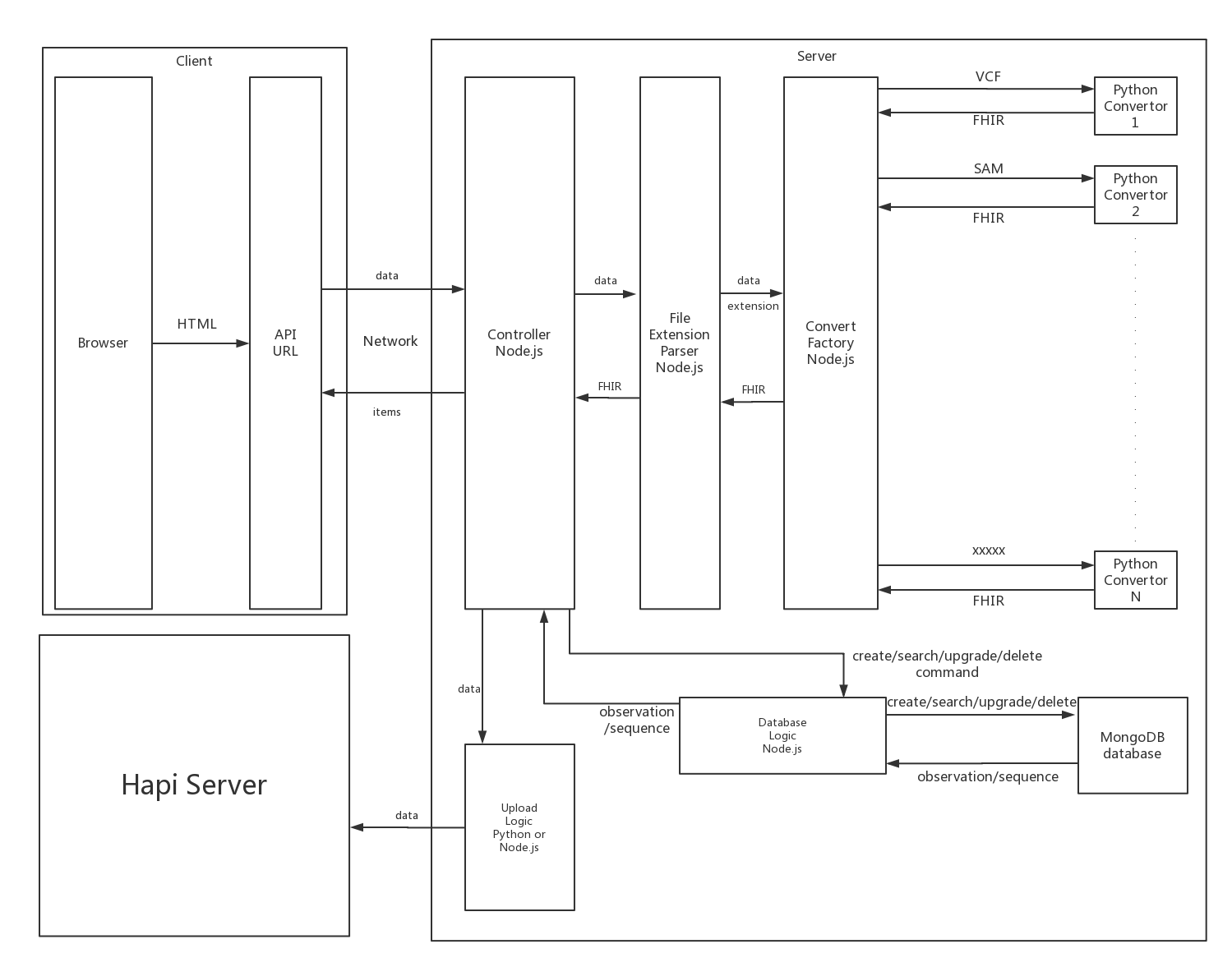
图1 系统架构
整个系统分为3部分,Client、Server和第三方的Hapi Server。对于Client来说,和大多数系统一样,我使用Restful API定义与Server的交互,并通过浏览器提供图形访问界面。而对于Server,有两部分,一部分用Node.js编写,一部分用python编写。Node.js主要负责业务逻辑的控制,Python负责编写脚本对上传的文件进行解析。而由于Hapi Server支持Python而暂时无第三方库支持Node.js,所以可以使用Python脚本执行上传的操作。整个业务流程是这样的 上传数据->Node.js接受并调用响应的Python脚本解析->存储到数据库中->用户增删改查或者按需指定记录上传->Node.js读取数据库并调用Python脚本将数据上传至Hapi Server。
在进一步的细化设计中,我使用了一些中间层进行解耦,使得Controller只需要负责控制,而将详细的业务逻辑解耦到具体的模块之中,使用Node.js进行控制模块之间的调用。
对于文件解析器,我们使用2个Node模块来实现功能——File Extension Parser和Convert Factory。我们使用工厂模式设计解析器,使得其可以方便进行扩展。当一个VCF文件上传到服务器时,Controller获取文件然后递交给File Extension Parser。File Extension Parser设计用来解析文件的后缀,并验证文件是否符合规范。之后将文件后缀传递至Convert Factory。Convert Factory获得文件的后缀并决定调用哪个Python脚本进行解析。顺便一说的是在File Extension中并没有选择解析器的业务逻辑。这样的设计实现了控制和功能的解耦,方便以后的改进以及扩展,类图如下:
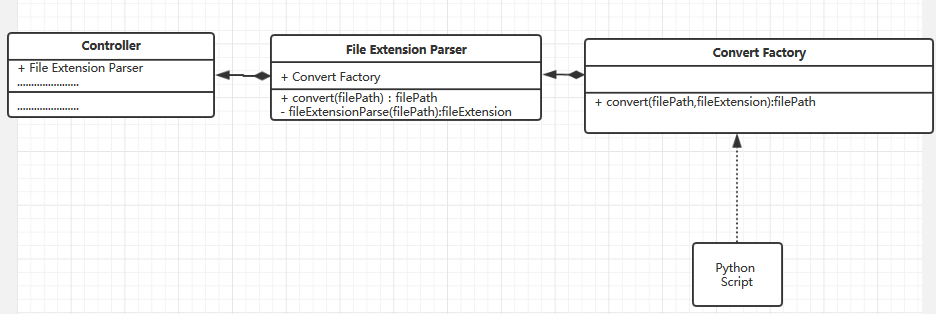
图2 Convertor类图
对于数据库层,我们优先选择的是非关系型数据库MongoDB,因为其能适应变化很多的数据格式。但如果后期要增加对HDFS、关系型数据库的支持,如果将数据库操作耦合到Controller中显然是不合适的,因此我们使用一层DataBase Logic的模块对系统进行解耦,将控制和功能分离。我们使用DataBase Logic模块来管理数据库的差异变化,并对Controller提供相同的接口。
对于Hapi Server的上传模块,我们也因为相同原因进行解耦。
三、心得体会
在这一次已经完成的实习中,有2个方面的感想体会:
一是对于新技术的探索。在这次的科研实习中,接触了两方面的新技术,一个是Node.js,一个是非关系数据库MongoDB。对于Node.js,其与我们一直使用的Java不一样,其是事件驱动的语言。而在对Node.js的学习中,我曾经对其事件轮询机制非常困惑,网上一方面说Node.js是单线程的服务器,一方面又说是异步I/O,这对我来说是非常迷惑的。经过不断地查阅资料和阅读文章,回忆操作系统的知识,明白了其事件轮询是单线程的,但对于每一个I/O操作又会单独开一个新的线程执行,最后用回调函数将结果返回给事件轮询的主线程。这个操作和PHP不一样的是,PHP为每一个请求开辟一个子线程,而Node.js则是用主线程接受请求,如果在响应请求的时候使用到了I/O操作,则会为其开启新的子线程,这样就节约了大量的在子线程中等待I/O操作的时间和资源。而对于MongoBD则为我打开了眼界,虽然上大数据课程的时候也已经知道,但真的操作的时候才发现其与我们之前学的关系型数据库不一样,崭新的体验。在这次的学习中,我发现了像操作系统之类的基本功的重要性,虽然并不会直接的做出来东西,但是不变应万变的内功心法。
二是对生物信息的学习。生物信息这门学科是我的初次尝试,其也是生物与计算机的交叉学科,但感觉更多的是使用计算机作为管理数据的方式,再加上一些机器学习的算法进行基因配对等任务。直观的感觉是这个领域标准不统一,挺复杂的,需要学习非常多的东西,而且这个学科目前的发展还是没有其他学科那么成熟,还需要非常多的人才去发展这个学科。但目前深度学习技术的兴起确实给这个学科带去了非常多的方法去做之前没办法做的事情,例如医疗图像检测等等,随着未来技术的进步,这个领域肯定能成为未来最火的领域,毕竟“21世纪是生物的世纪”。
 文档下载
文档下载 会议室预约
会议室预约 管理登录
管理登录 EN
EN 会议室预约
会议室预约 管理登录
管理登录
